| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||



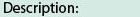 Catalytic domain of Myotonic Dystrophy protein kinase-like Protein Serine/Threonine Kinases. Serine/Threonine Kinases (STKs), Myotonic Dystrophy protein kinase (DMPK)-like subfamily, catalytic (c) domain. STKs catalyze the transfer of the gamma-phosphoryl group from ATP to serine/threonine residues on protein substrates. The DMPK-like subfamily is part of a larger superfamily that includes the catalytic domains of other protein STKs, protein tyrosine kinases, RIO kinases, aminoglycoside phosphotransferase, choline kinase, and phosphoinositide 3-kinase. The DMPK-like subfamily is composed of DMPK and DMPK-related cell division control protein 42 (Cdc42) binding kinase (MRCK). Three isoforms of MRCK are known, named alpha, beta and gamma. The DMPK gene is implicated in myotonic dystrophy 1 (DM1), an inherited multisystemic disorder with symptoms that include muscle hyperexcitability, progressive muscle weakness and wasting, cataract development, testicular atrophy, and cardiac conduction defects. The genetic basis for DM1 is the mutational expansion of a CTG repeat in the 3'-UTR of DMPK. DMPK is expressed in skeletal and cardiac muscles, and in central nervous tissues. The functional role of DMPK is not fully understood. It may play a role in the signal transduction and homeostasis of calcium. MRCK is activated via interaction with the small GTPase Cdc42. MRCK/Cdc42 signaling mediates myosin-dependent cell motility. MRCKgamma is expressed in heart and skeletal muscles, unlike MRCKalpha and MRCKbeta, which are expressed ubiquitously.
Catalytic domain of Myotonic Dystrophy protein kinase-like Protein Serine/Threonine Kinases. Serine/Threonine Kinases (STKs), Myotonic Dystrophy protein kinase (DMPK)-like subfamily, catalytic (c) domain. STKs catalyze the transfer of the gamma-phosphoryl group from ATP to serine/threonine residues on protein substrates. The DMPK-like subfamily is part of a larger superfamily that includes the catalytic domains of other protein STKs, protein tyrosine kinases, RIO kinases, aminoglycoside phosphotransferase, choline kinase, and phosphoinositide 3-kinase. The DMPK-like subfamily is composed of DMPK and DMPK-related cell division control protein 42 (Cdc42) binding kinase (MRCK). Three isoforms of MRCK are known, named alpha, beta and gamma. The DMPK gene is implicated in myotonic dystrophy 1 (DM1), an inherited multisystemic disorder with symptoms that include muscle hyperexcitability, progressive muscle weakness and wasting, cataract development, testicular atrophy, and cardiac conduction defects. The genetic basis for DM1 is the mutational expansion of a CTG repeat in the 3'-UTR of DMPK. DMPK is expressed in skeletal and cardiac muscles, and in central nervous tissues. The functional role of DMPK is not fully understood. It may play a role in the signal transduction and homeostasis of calcium. MRCK is activated via interaction with the small GTPase Cdc42. MRCK/Cdc42 signaling mediates myosin-dependent cell motility. MRCKgamma is expressed in heart and skeletal muscles, unlike MRCKalpha and MRCKbeta, which are expressed ubiquitously. No pairwise interactions are available for this conserved domain.
No pairwise interactions are available for this conserved domain.



Tips:  Range on the Protein: Protein ID Protein Position Domain Position: 

|
|---|



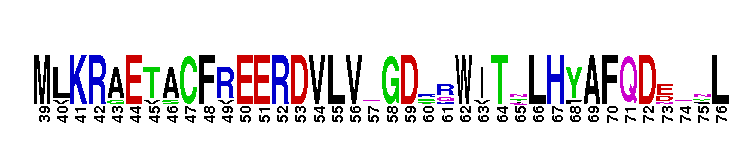
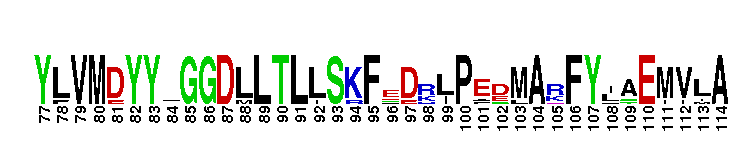




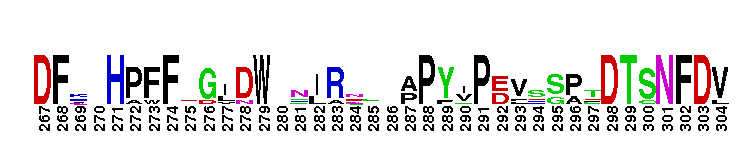

Weblogos are Copyright (c) 2002 Regents of the University of California
| DMDM_info@umbc.edu | 1000 Hilltop Circle, Baltimore, MD 21250 | Department of Biological Sciences | Phone: 410-455-2258 |